Quand vous prenez un médicament générique, comment savez-vous qu’il va agir exactement comme le médicament de marque ? La réponse se trouve dans deux concepts clés de la pharmacologie : la biodisponibilité et l’équivalence biologique. Beaucoup les confondent, mais ils ne sont pas la même chose. L’un décrit comment un seul produit est absorbé par le corps. L’autre compare deux produits pour vérifier s’ils sont interchangeables. Comprendre la différence est essentiel, surtout si vous ou un proche prenez des traitements à indice thérapeutique étroit, comme la warfarine ou la lévothyroxine.
Qu’est-ce que la biodisponibilité ?
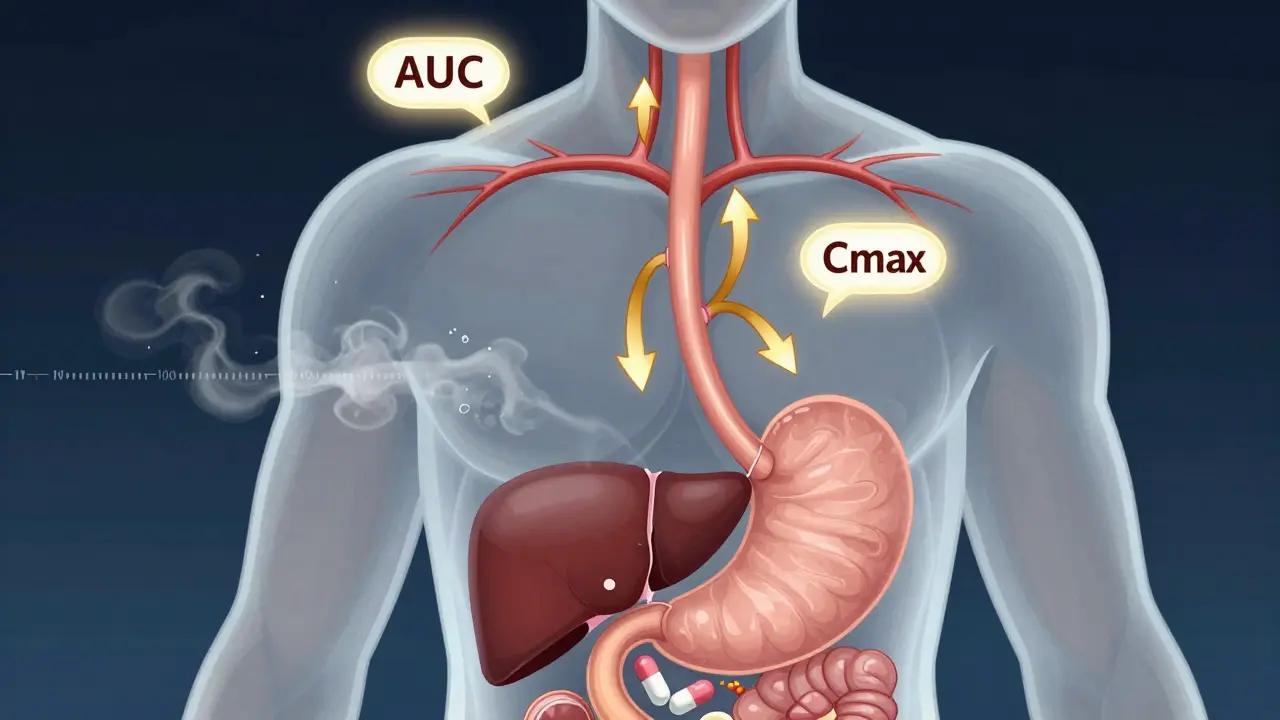
La biodisponibilité mesure la quantité de médicament qui atteint la circulation sanguine après administration. Elle répond à une question simple : combien de la dose que vous avalez, inhalez ou injectez arrive vraiment là où elle doit agir ?
Pour un médicament pris par voie orale, ce n’est pas 100 %. Une partie est perdue dans l’estomac, une autre détruite par le foie avant même d’atteindre le sang - on appelle ça le passage de premier passage. Par exemple, si un médicament a une biodisponibilité de 50 %, cela signifie que seulement la moitié de la dose ingérée entre en circulation. Le reste est éliminé avant d’agir.
On mesure la biodisponibilité avec deux paramètres clés : l’AUC (aire sous la courbe concentration-temps) et la Cmax (concentration maximale dans le sang). L’AUC indique la quantité totale de médicament absorbée sur une période. La Cmax montre à quel point et à quelle vitesse le médicament atteint son pic d’efficacité. Pour les médicaments injectés par voie intraveineuse, la biodisponibilité est toujours de 100 % - c’est la référence absolue. Pour les autres formes (comprimés, gélules, sirops), on compare leur AUC et Cmax à celle de l’IV pour déterminer leur biodisponibilité absolue.
La biodisponibilité relative, elle, compare deux formulations du même médicament. Par exemple, un comprimé générique contre le comprimé de marque. Ce n’est pas une mesure isolée : c’est toujours un rapport entre deux produits.
Qu’est-ce que l’équivalence biologique ?
L’équivalence biologique, c’est la vérification que deux produits contenant le même principe actif - généralement un générique et son original - produisent des effets thérapeutiques identiques. Ce n’est pas une hypothèse. C’est une preuve scientifique exigeante.
Pour l’établir, les autorités sanitaires exigent une étude clinique comparée : 24 à 36 volontaires en bonne santé prennent les deux médicaments, dans un ordre aléatoire, avec un intervalle de lavage entre les prises. On prélève leur sang à 12 à 18 moments précis sur 72 heures. On calcule l’AUC et la Cmax pour chaque produit, puis on compare les résultats.
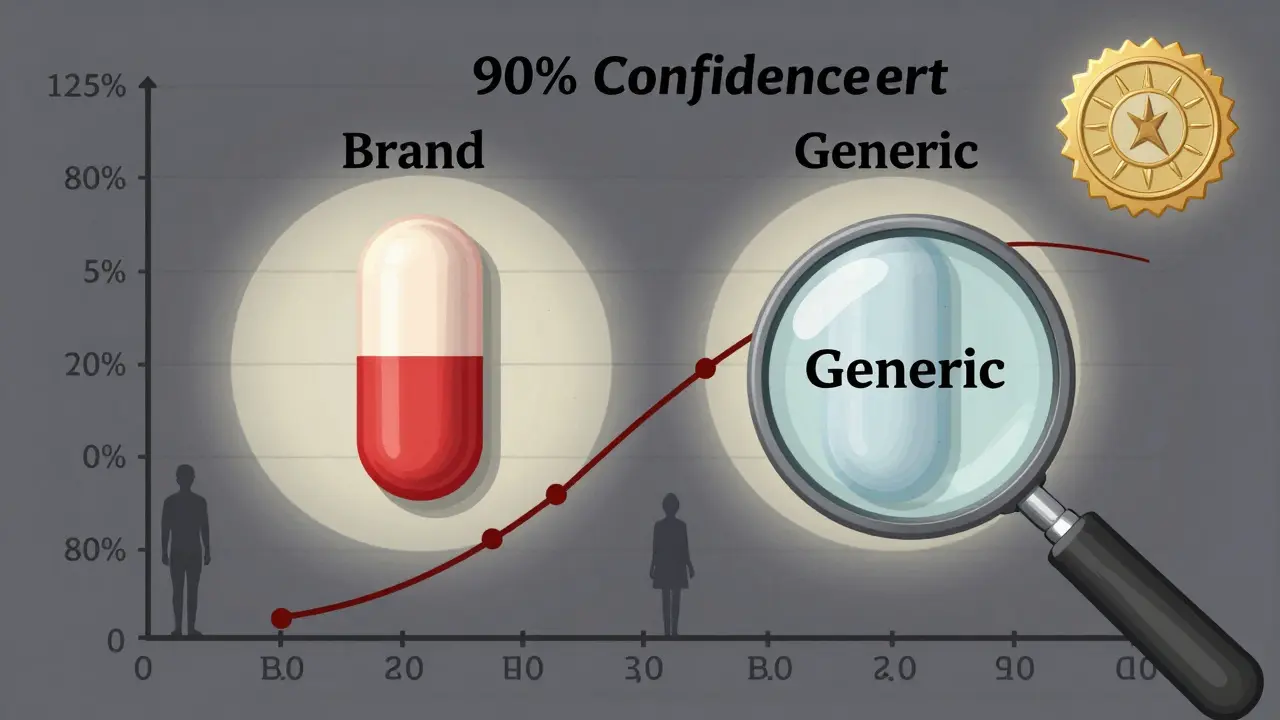
La règle de base ? La plage de confiance à 90 % du rapport des moyennes géométriques de l’AUC et de la Cmax doit se situer entre 80 % et 125 %. C’est ce qu’on appelle la règle 80/125. Si le générique a une AUC à 92 % de l’original, et une Cmax à 118 %, il est équivalent. Si l’un des deux sort de cette plage, il n’est pas approuvé.
Pourquoi cette plage ? Parce que les réponses pharmacocinétiques ne suivent pas une logique additive, mais multiplicative. Une différence de 20 % en valeur absolue n’est pas la même chose qu’une différence de 20 % en ratio. La règle 80/125 a remplacé l’ancien modèle ±20 % dans les années 1990 pour cette raison. Elle est plus précise, plus juste, et elle a fait ses preuves.
La différence fondamentale entre les deux concepts
La biodisponibilité est une propriété d’un seul produit. Elle dit : « Ce comprimé, à lui seul, libère 60 % de son principe actif dans le sang. »
L’équivalence biologique est une comparaison entre deux produits. Elle dit : « Ce générique libère entre 80 % et 125 % de ce que libère le médicament de référence. »
Autrement dit, vous pouvez mesurer la biodisponibilité d’un seul médicament. Mais vous ne pouvez pas parler d’équivalence biologique sans avoir deux produits à comparer. L’équivalence biologique repose sur la biodisponibilité, mais elle va plus loin : elle ajoute une norme statistique, une méthode d’étude, et une exigence réglementaire.
Par exemple, un médicament peut avoir une biodisponibilité de 45 % - c’est faible, mais ce n’est pas un problème en soi. Ce qui compte, c’est que le générique ait une biodisponibilité entre 36 % et 56,25 % (45 % × 0,8 à 45 % × 1,25). Si c’est le cas, il est équivalent.
Des exceptions : les médicaments à indice thérapeutique étroit
La règle 80/125 marche bien pour la plupart des médicaments. Mais pour certains, même une petite variation peut être dangereuse. Ce sont les médicaments à indice thérapeutique étroit (ITÉ) : la différence entre une dose efficace et une dose toxique est mince.
La warfarine, la lévothyroxine, la phénytoïne, la ciclosporine : ces traitements exigent une précision extrême. Pour eux, la FDA a resserré les limites. Pour la warfarine, la plage d’acceptation pour l’AUC est de 90 % à 112 %, et non 80 % à 125 %. Pour la Cmax, elle reste à 80/125, car les pics de concentration sont moins critiques que l’exposition totale.
Des études montrent que les patients passant d’un générique à un autre de la lévothyroxine, même si tous sont « équivalents » selon la FDA, rapportent parfois des variations de leur TSH. Cela ne signifie pas que les normes sont fausses. Cela signifie que pour ces médicaments, les différences de formulation (excipients, granulométrie, taux de dissolution) peuvent avoir un impact plus sensible que prévu.
En 2022, une étude publiée dans Clinical Pharmacology & Therapeutics a montré que 37 % des patients ayant changé de générique de lévothyroxine ont eu une variation de TSH supérieure à 20 %. Seuls 12 % ont eu des symptômes cliniques, mais cela suffit pour alerter les prescripteurs. Certains médecins préfèrent garder le même générique une fois qu’il fonctionne bien.
Comment les études sont-elles réalisées ?
Une étude d’équivalence biologique n’est pas une simple comparaison de prix. C’est une opération scientifique complexe.
- Les volontaires sont en jeûne (pour éliminer l’effet des aliments).
- Les prélèvements sanguins sont fréquents : toutes les 15 à 60 minutes pendant 72 heures.
- Les analyses sont faites par chromatographie en phase liquide avec spectrométrie de masse - une technique très précise.
- Les données sont transformées en logarithme avant d’être analysées statistiquement.
- Les résultats doivent être reproduits dans au moins deux études indépendantes.
Un seul problème peut bloquer l’approbation : un effet de repas. Par exemple, le voriconazole - un antifongique - voit sa Cmax augmenter de 36 % et son AUC de 23 % après un repas gras. Pour ce médicament, il faut mener deux études : l’une à jeun, l’autre après un repas. Le générique doit être équivalent dans les deux cas.
Les laboratoires de recherche contractuelle (CRO) mettent entre 3 et 6 mois pour concevoir ces études. Les coûts peuvent atteindre plusieurs centaines de milliers d’euros. C’est pourquoi les grands fabricants de génériques investissent massivement dans ces tests : ils savent que sans équivalence, pas d’approbation, pas de vente.

Les résultats dans le monde réel
Les chiffres sont rassurants. Entre 2010 et 2020, 99,7 % des génériques approuvés par la FDA ont passé l’épreuve de l’équivalence biologique. Aux États-Unis, 91 % des ordonnances sont remplies par des génériques, mais ils ne représentent que 22 % des dépenses totales en médicaments.
Une étude menée par l’Association américaine des pharmaciens a suivi 1 247 patients passés du médicament de marque au générique pour l’hypertension. Seuls 17 ont signalé des problèmes. Après vérification, seulement 4 cas étaient réellement liés à une différence d’absorption. Les autres étaient dus à une mauvaise adhérence, à des variations de l’état de santé, ou à des effets placebo.
Pourtant, des patients sur Reddit et des groupes de défense comme Patients for Better Drugs rapportent des cas où les génériques semblent moins efficaces. La plupart du temps, ces différences ne sont pas prouvées scientifiquement. Mais elles existent. Et elles ne doivent pas être ignorées.
Les experts s’accordent sur un point : l’équivalence biologique est un système robuste. Elle a permis à des millions de patients d’accéder à des traitements abordables sans compromettre la sécurité. Mais elle n’est pas parfaite. Pour les ITÉ, il faut rester vigilant, surveiller les marqueurs biologiques, et parfois privilégier la constance du générique plutôt que la réduction de coût.
Et demain ?
Les normes évoluent. La FDA travaille sur des lignes directrices spécifiques pour les génériques complexes : pommades, inhalateurs, produits injectables à liposomes. Pour ces produits, les paramètres sanguins traditionnels ne suffisent pas. Des méthodes alternatives sont en cours d’élaboration, comme les tests de dissolution in vitro ou la modélisation pharmacocinétique basée sur la physiologie (PBPK).
En 2024, l’Union européenne envisage d’autoriser des études de dissolution pour remplacer partiellement les études sur les volontaires, si les données sont suffisamment solides. Cela pourrait réduire les coûts et accélérer l’accès aux génériques.
Le marché mondial des tests d’équivalence biologique devrait atteindre 4,5 milliards de dollars d’ici 2027. Ce n’est pas juste un secteur technique - c’est un pilier de la santé publique.
En fin de compte, comparer la biodisponibilité et l’équivalence biologique, c’est comprendre que la science derrière un simple comprimé générique est immense. Ce n’est pas une copie. C’est une réplique rigoureusement validée. Et c’est ce qui permet à des millions de personnes de vivre plus longtemps, mieux, et à moindre coût.
Quelle est la différence entre biodisponibilité et équivalence biologique ?
La biodisponibilité mesure la quantité et la vitesse à laquelle un seul médicament est absorbé par l’organisme. L’équivalence biologique compare deux médicaments (généralement un générique et un original) pour vérifier qu’ils ont des profils d’absorption similaires, selon des critères statistiques stricts (plage de 80-125 % pour l’AUC et la Cmax).
Pourquoi la FDA utilise-t-elle la plage 80-125 % pour l’équivalence biologique ?
Cette plage garantit que la différence d’exposition entre deux produits ne dépasse pas 20 % dans les deux sens, ce qui est considéré comme cliniquement négligeable pour la majorité des médicaments. Elle repose sur des données pharmacocinétiques et statistiques, et elle a remplacé l’ancien modèle ±20 % car les réponses biologiques suivent une logique multiplicative, pas additive.
Tous les médicaments génériques sont-ils équivalents à leurs versions de marque ?
Oui, selon les normes réglementaires. Tous les génériques approuvés par la FDA, l’EMA ou d’autres agences doivent démontrer une équivalence biologique. Cependant, pour les médicaments à indice thérapeutique étroit (comme la lévothyroxine), de rares patients peuvent ressentir des différences dues à des variations de formulation. Dans ces cas, il est recommandé de rester sur le même générique une fois stabilisé.
Les études d’équivalence biologique sont-elles fiables ?
Oui, elles sont très fiables. Elles sont réalisées dans des conditions contrôlées, avec des volontaires sains, des prélèvements sanguins fréquents et des analyses de haute précision. Les données sont analysées selon des protocoles internationaux. La fiabilité est confirmée par des décennies d’utilisation clinique : moins de 0,3 % des patients signalent des échecs thérapeutiques liés à l’équivalence biologique.
Pourquoi certains patients disent-ils que les génériques ne marchent pas aussi bien ?
Les raisons sont souvent non liées à la biodisponibilité. Elles peuvent inclure des changements dans les excipients (qui affectent la perception du médicament), des variations de l’état de santé, une mauvaise prise du traitement, ou l’effet placebo/nocebo. Dans les cas rares où une différence réelle est confirmée, elle concerne principalement les médicaments à indice thérapeutique étroit, et les autorités adaptent alors les seuils d’équivalence.


Suzanne Brouillette
décembre 27, 2025 AT 00:38C’est fou comment on pense que c’est juste du plastique et du lactose, mais en fait, c’est un chef-d’œuvre de pharmacocinétique 😊
Elise Alber
décembre 28, 2025 AT 04:38La biodisponibilité relative est souvent mal comprise : c’est un ratio, pas une différence absolue. Et la règle 80-125 ? Elle repose sur une logique multiplicative, pas additive - un détail que beaucoup d’interprètes négligent, surtout dans les revues non spécialisées.
La Cmax et l’AUC ne sont pas des valeurs isolées : leur variabilité inter-individuelle peut atteindre 30-40 % même avec le même produit. C’est pourquoi les études utilisent des modèles linéaires mixtes avec transformation log. Et oui, les excipients influencent la dissolution - surtout avec les formulations à libération modifiée.
Les études sur volontaires sains ne reflètent pas toujours la population réelle : âge, comorbidités, polymorphismes CYP450, interactions médicamenteuses… tout ça change la donne. Pourtant, on continue à appliquer la même norme à la warfarine et à l’ibuprofène.
La FDA a raison de resserrer les seuils pour les ITÉ, mais pourquoi ne pas exiger des études post-marketing obligatoires pour ces cas ? On attend que les patients se plaignent pour réagir…
Et puis, la question des génériques de lévothyroxine : 37 % de variations de TSH ? C’est énorme. Mais les médecins refusent de l’admettre parce que ça remet en cause leur confiance dans les agences. Le système est robuste - mais pas infallible.
Le vrai problème, c’est la transparence : les protocoles des études d’équivalence ne sont pas publiés. On ne sait pas comment les CRO ont traité les données, ni quelles exclusions ont été faites. Sans ça, c’est de la foi, pas de la science.
Je veux bien croire que 99,7 % des génériques sont équivalents - mais comment vérifier ça sans accès aux données brutes ? La régulation est un black box.
Et les patients qui disent « ça ne marche pas » ? Ils ne sont pas des cons. Ils ressentent des différences. Peut-être que la norme 80-125 est trop large pour certains profils pharmacogénétiques.
Je ne dis pas qu’il faut abandonner les génériques. Je dis qu’il faut les surveiller avec plus de rigueur - pas juste les approuver et oublier.
Jérémy Dabel
décembre 29, 2025 AT 23:56moi j'ai changé de genrique de levothyroxine et j'ai eu des palpitations... j'ai retourne a l'ancien et tout s'est calme. j'peux pas dire que c'est la science qui a tort, mais j'peux dire que mon corps a senti la difference.
Guillaume Franssen
décembre 31, 2025 AT 09:52Ok mais sérieux, pourquoi on ne parle JAMAIS de la qualité des excipients ?!?!?!
Un générique, c’est pas juste le principe actif - c’est aussi le colorant, le liant, le taux d’humidité, la granulométrie…
Et si le produit de marque utilise du lactose purifié à 99,9 %, et que le générique utilise du lactose industriel avec des résidus de protéines de lait ?
Ça change rien pour 99 % des gens… mais pour les intolérants ? Ou les hyper-sensibles ?
Et personne ne teste ça en profondeur !
On se contente de mesurer la Cmax et l’AUC… mais si ton corps réagit aux impuretés, c’est pas de la biodisponibilité, c’est de la toxicité subclinique !
Et on appelle ça de la « science » ?!?!
Je suis pharmacien, j’ai vu des patients qui avaient des réactions à chaque changement de générique… et les agences disent : « c’est psychologique ».
Non. C’est qu’on a pas encore les outils pour mesurer tout ça. Et c’est pas une excuse.
Élaine Bégin
janvier 1, 2026 AT 06:10Arrêtez de faire des crises de nerfs pour un comprimé. Si votre TSH bouge de 20 %, c’est que vous ne prenez pas votre médicament à la même heure, ou que vous mangez du soja le matin. C’est pas le générique qui est en cause, c’est vous.
Jean-François Bernet
janvier 1, 2026 AT 07:51La règle 80-125 ? C’est une farce. Une blague de laboratoire pour justifier que les multinationales puissent vendre des copies à 10 % du prix. Vous croyez vraiment que deux comprimés avec des excipients différents ont le même effet ?
Regardez les études : 24 volontaires en bonne santé. 24. Pas 24 000. Pas de diabétiques. Pas de seniors. Pas de patients avec des foies fatigués.
Et vous appelez ça « équivalence » ?
Je vous dis : c’est de la manipulation. Et les médecins qui répètent ça comme des perroquets ? Des complices.
Cassandra Hans
janvier 2, 2026 AT 21:31Vous parlez de biodisponibilité… mais qui a vérifié la stabilité du générique après 18 mois de stockage dans une armoire humide ?
Qui a mesuré la dissolution dans un environnement gastrique acide à pH 1,8 vs 2,5 ?
Qui a testé l’impact des variations de température pendant le transport ?
La réglementation se contente de « l’AUC et la Cmax »… mais la vraie biodisponibilité, c’est ce que le corps reçoit, pas ce que le laboratoire affirme.
Et puis… pourquoi les génériques ont-ils souvent un goût différent ? Parce que les excipients changent. Et le goût, ça influence l’adhérence. Et l’adhérence, ça influence l’efficacité.
On ne peut pas réduire la santé à deux chiffres statistiques.
On a perdu la notion de complexité biologique.
Caroline Vignal
janvier 4, 2026 AT 13:19STOP. Le générique, c’est la liberté. La liberté de vivre sans être ruiné. La liberté de prendre son traitement tous les jours. La liberté de ne pas choisir entre manger et médicaments.
Si vous avez un problème avec un générique ? Changez-en. Parlez à votre pharmacien. Mais ne dénigrez pas un système qui sauve des vies à chaque instant.
99,7 % de réussite. C’est plus que la plupart des choses dans la vie.
Et si vous voulez du parfait ? Payez 500€ le comprimé.
Je choisis la vie. Pas la perfection.
olivier nzombo
janvier 5, 2026 AT 05:34Je suis un patient de la lévothyroxine depuis 12 ans. J’ai changé 4 fois de générique. Chaque fois, j’ai eu des symptômes : fatigue, dépression, prise de poids. J’ai fait des analyses. TSH à 7, puis 4, puis 8. Chaque fois, je suis revenu à l’ancien. Et tout allait mieux.
La science dit que c’est impossible. Mon corps dit le contraire.
Je ne suis pas un cas isolé. Je suis une voix parmi des milliers.
La réglementation est une machine froide. Mais les corps humains… ils ne sont pas des chiffres.
Je veux un générique. Mais je veux aussi que quelqu’un m’écoute.
Et non, je ne suis pas parano. Je suis vivant.
Raissa P
janvier 7, 2026 AT 02:07La biodisponibilité est une illusion. L’équivalence, une illusion plus grande.
Tout est relation. Tout est contexte. Un médicament n’est pas une chose. C’est un événement entre le corps, le monde, le temps.
Vous mesurez la Cmax ? Mais vous ne mesurez pas la douleur. Vous mesurez l’AUC ? Mais vous ne mesurez pas l’angoisse.
La science veut réduire la vie à des courbes. Mais la vie ne suit pas les courbes.
Elle suit les rythmes. Les saisons. Les émotions. Les silences.
Et quand un patient dit « ça ne marche plus », ce n’est pas un échec de la pharmacie. C’est un appel à l’humilité.
James Richmond
janvier 7, 2026 AT 23:56Les gens qui disent que les génériques sont mauvais… ils ont juste peur de changer. C’est comme changer de téléphone. Tu penses que le nouveau est pire, mais c’est juste que t’es pas habitué.
La science a prouvé que c’est pareil. Arrête de croire aux histoires de ton voisin.
theresa nathalie
janvier 9, 2026 AT 02:01Je suis pharmacienne. J’ai vu des patients qui avaient des réactions à chaque changement de générique. J’ai vu des TSH qui montaient en flèche. J’ai vu des gens qui pleuraient parce qu’ils ne comprenaient pas pourquoi ils se sentaient mal… alors que tout était « dans les normes ».
Je ne dis pas que les génériques sont dangereux.
Je dis qu’on devrait leur donner un nom différent. Pas juste « générique ». Un nom qui dit : « ce n’est pas exactement le même ». Parce que ce n’est pas exactement le même.
Et si on arrêtait de dire « c’est psychologique » et qu’on commençait à écouter ?
Suzanne Brouillette
janvier 10, 2026 AT 03:02Je suis contente que vous parliez de ça. J’ai un patient qui a changé de générique et a eu une crise d’angine… on a retrouvé un pic de concentration à 132 % de Cmax. Le générique était dans les normes, mais pour lui, c’était trop. On l’a réadapté. La science est une base. Mais la pratique, c’est l’art de l’ajustement. ❤️