
Vous avez déjà entendu parler des médicaments génériques, ces alternatives moins chères qui ont révolutionné l'accès aux soins. Mais saviez-vous que pour les produits combinés des formulations contenant plusieurs principes actifs ou associant un médicament à un dispositif médical, la démonstration de leur équivalence est bien plus complexe ? Ce n'est pas une simple question de copier une molécule unique. Il s'agit de prouver que chaque composant fonctionne exactement comme dans le produit original, sans interagir de manière imprévisible.
Aujourd'hui, environ 73 % des nouvelles entités chimiques approuvées sont des produits complexes. Pourtant, les obstacles réglementaires ralentissent considérablement l'arrivée des génériques sur le marché. Pourquoi est-ce si difficile ? Et comment les laboratoires parviennent-ils à surmonter ces défis techniques ? Découvrons ensemble les enjeux cachés derrière ces tests cruciaux.
Qu'est-ce qu'un produit combiné complexe ?
Dans le langage courant, on pense souvent à un simple comprimé. Mais un produit combiné va bien au-delà. Il peut s'agir d'une association fixe de deux principes actifs (comme certains traitements contre le VIH), d'une crème dermatologique complexe, ou encore d'un dispositif médical intégrant un médicament, comme un inhalateur pour l'asthme.
L'Agence européenne des médicaments (EMA) l'agence chargée d'évaluer et de surveiller les médicaments dans l'Union européenne et la Food and Drug Administration (FDA) l'autorité de régulation des États-Unis pour les aliments et les médicaments définissent strictement ces produits. Le but est d'assurer que le générique délivre les substances actives au même rythme et dans la même mesure que le produit de référence. Cependant, quand plusieurs ingrédients interagissent, cette tâche devient un casse-tête scientifique.
Les défis des associations fixes de doses (FDC)
Les associations fixes de doses, ou FDC, posent un problème majeur : les interactions entre les principes actifs. Imaginez mélanger deux liquides qui réagissent chimiquement ; le résultat n'est pas toujours prévisible. Dans le corps humain, si une molécule insoluble est associée à une autre, elle peut modifier l'absorption de son partenaire.
Pour contourner cela, les développeurs doivent prouver la bioéquivalence par rapport aux produits mono-molécules individuels ET par rapport au produit combiné de référence. Cela nécessite des protocoles croisés en trois voies, augmentant la puissance statistique mais aussi la complexité. Selon les données de la FDA, les taux d'échec initiaux pour ces études peuvent atteindre 35 à 40 %, bien plus élevés que pour les médicaments simples.
- Taille de l'échantillon : Alors qu'un test standard nécessite 24 à 36 volontaires, un FDC en demande souvent 40 à 60.
- Analyse multiple : Il faut mesurer simultanément plusieurs analytes, ce qui complique l'interprétation des résultats.
- Coût accru : Ces études représentent 30 à 40 % du budget total de développement, soit des millions de dollars supplémentaires.
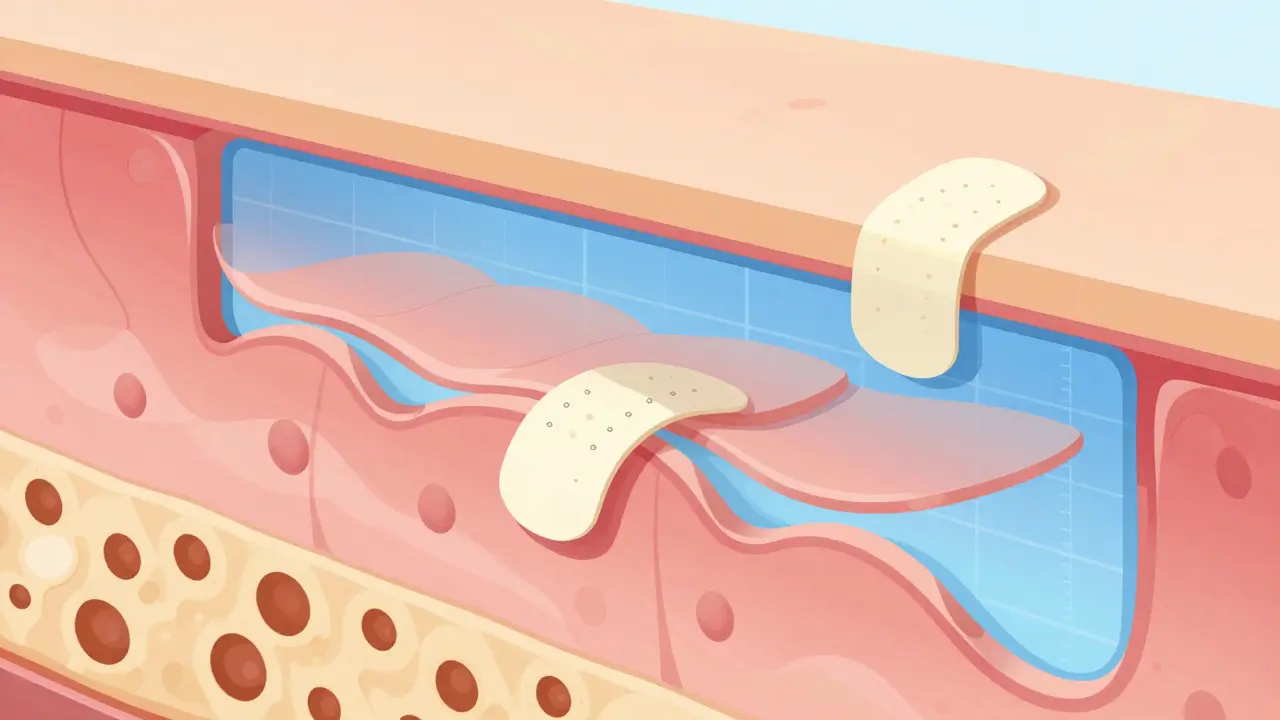
La difficulté des produits topiques et dermiques
Lorsqu'il s'agit de crèmes ou de pommades, le défi change de nature. Comment quantifier précisément la quantité de médicament qui pénètre dans la couche cornée de la peau ? Les méthodes actuelles reposent sur des procédures de "tape-stripping", où l'on prélève 15 à 20 couches successives de la peau.
Le problème ? Il n'existe pas de spécifications claires sur la profondeur exacte ou la quantité à analyser. Cette ambiguïté crée une grande variabilité entre les laboratoires. Une étude publiée dans Frontiers in Pharmacology en 2024 souligne cette lacune. Par conséquent, les études comparatives sur les points finaux cliniques deviennent nécessaires, impliquant 200 à 300 patients par bras d'étude. Le coût grimpe alors à 5-10 millions de dollars, rendant ces approches peu rentables pour de nombreux fabricants de génériques.
Produits combinés médicament-dispositif (DDCP)
Les dispositifs médicaux intégrés, comme les inhalateurs doseurs ou les injecteurs, introduisent une variable humaine critique : l'interface utilisateur. Une légère différence de design peut radicalement changer la façon dont le patient utilise le dispositif, affectant ainsi la livraison du médicament.
Dr. William Doub de la FDA a déclaré lors d'un atelier en 2024 que l'évaluation comparative de l'interface utilisateur reste le plus grand obstacle pour l'approbation des DDCP génériques. En effet, 65 % des lettres de réponse complète citent des carences dans ce domaine. Pour les inhalateurs, il faut vérifier que la distribution de la taille des particules aérodynamiques se situe entre 80 % et 120 % des performances du produit de référence. Une marge étroite qui demande une ingénierie de précision.
| Type de Produit | Défis Principaux | Taille Échantillon Type | Coût Estimé Étude |
|---|---|---|---|
| Médicament Oral Simple | Absorption basique | 24-36 sujets | 1-2 millions $ |
| Association Fixe (FDC) | Interactions API, analyse multiple | 40-60 sujets | 15-25 millions $ (total dev.) |
| Topique/Dermique | Pénétration cutanée, variabilité | 200-300 patients (clinique) | 5-10 millions $ |
| Médicament-Dispositif | Interface utilisateur, performance | Variable | Élevé (tests spécifiques) |

Solutions innovantes et modélisation
Face à ces coûts prohibitifs, l'industrie cherche des raccourcis intelligents. La modélisation pharmacocinétique physiologiquement basée (PBPK) gagne du terrain. Elle permet de simuler le comportement du médicament dans le corps virtuel, réduisant le besoin d'études cliniques massives.
Jusqu'à la deuxième trimestre 2024, la FDA a accepté cette approche dans 17 demandes d'autorisation de mise sur le marché (ANDA) pour des produits complexes. Cela réduit les études cliniques requises de 30 à 50 %. De plus, le Consortium des Produits Complexes de la FDA, créé en 2021, a développé 12 recommandations spécifiques, permettant de réduire les délais de développement de 8 à 12 mois pour les entreprises participantes.
Les réunions de type II avec la FDA, qui permettent de discuter du plan d'étude avant lancement, ont augmenté de 220 % depuis 2020. C'est un signe clair que l'industrie cherche activement à clarifier ces chemins réglementaires flous.
Impact économique et avenir réglementaire
L'enjeu dépasse la science pure. Les médicaments génériques ont économisé 373 milliards de dollars au système de santé américain en 2020 seul. Pourtant, les produits complexes restent protégés par des "épaisseurs de brevets" qui retardent l'entrée des génériques de près de 2,3 ans en moyenne.
Le marché mondial des génériques complexes atteignait 112,7 milliards de dollars en 2023. Si les défis de bioéquivalence ne sont pas résolus, on estime que 45 % des produits de marque complexes resteront sans concurrence générique jusqu'en 2030. La FDA lance donc une "Initiative de Modernisation de la Bioéquivalence" visant à publier 50 nouvelles directives spécifiques d'ici 2027, en commençant par les produits respiratoires.
Pourquoi la bioéquivalence est-elle plus difficile pour les produits combinés ?
Parce que plusieurs composants peuvent interagir entre eux, modifiant leur absorption ou leur efficacité. De plus, pour les dispositifs, l'utilisation par le patient ajoute une variable imprévisible.
Qu'est-ce que le "tape-stripping" dans les tests topiques ?
C'est une méthode consistant à prélever des couches successives de la peau avec du ruban adhésif pour mesurer la quantité de médicament absorbée par la couche cornée.
Comment la modélisation PBPK aide-t-elle les développeurs ?
Elle permet de simuler numériquement le comportement du médicament, réduisant ainsi le nombre de patients nécessaires pour les essais cliniques réels et diminuant les coûts.
Quel est le coût moyen d'une étude de bioéquivalence pour un produit topique ?
Il peut varier entre 5 et 10 millions de dollars, principalement en raison de la nécessité d'inclure un grand nombre de patients (200-300) dans des études cliniques comparatives.
Que prévoit la FDA pour simplifier ces processus ?
La FDA vise à publier 50 nouvelles directives spécifiques aux produits complexes d'ici 2027 et encourage l'utilisation de modèles numériques et de standards de référence harmonisés.


